Design Process
In vitro modifications
In vivo modifications
Due to budget constraints, we tested our designs in vitro as DNA molecular beacons rather than using RNA. ssRNA sequences are significantly more expensive to purchase than ssDNA sequences, and given that many of our designs were novel, we felt it would be a better use of budget to test multiple switch designs.
The riboswitch designs that we tested in vitro were converted straight from RNA without a second Nupack sequence optimisation in DNA. Although more likely to cause issues with folding this was deemed important for the in vitro and in vivo testing to be comparable.
Unlike conventional molecular beacons, that are labelled with a fluorophore (Fl) and quencher (Q) on the ends of the stem, we attached 20nt 5’ and 3’ flanking sequences (in fact the lac operator sequence preceding our switch and the first segment of mKate2 from our in vivo designs). These flanking sequences are the same for all the switches and complementary to a universal set of 20nt labelled, DNA oligos. This enabled us to save money by ordering only one set of labelled oligonucleotides that would work for all probes:
The addition of strands to the solution can lead to structural changes in our switches. If this structural change separates the fluorophore and quencher, as should be the case in our activatory designs, then this will lead to an increase in fluorescence. If addition of a strand causes the fluorophore and quencher to get closer together then the fluorescence should decrease. This means that fluorescence can be used as a readout of the shape of our switches.
We used the fluorophore-quencher pair FAM and BHQ due to their low cost, high stability and good spectral overlap.
However to get a more direct measure of the accessibility of the RBS, the more significant readout in vivo, we ordered a short oligo that is complementary to the RBS sequence of our switch that has been labelled with a fluorophore. We called this the "Mimic" as it was intended to mimic the 16S rRNA of the small subunit of the prokaryotic ribosome that is complementary to the ribosome binding sequence.
Due to minimum length requirements of labelled-oligos the Mimic strand overlaps by two nucleotides with the quencher-labelled oligo binding sequence. We therefore made a new slightly extended version of the homo sym switch so that the mimic and quencher oligo could both bind in the open state when fluorescence should be low. In the closed structure however the rbs should be inaccessible and only the quencher can bind so fluorescence should be high.
To act as a non-cooperative control for activatory designs we ordered strands complementary to each of our activatory switch sequences, going up right up to the fluorophore and quencher annealed flanking sequences. These NC strands were added in excess as the final lane of our plate reader experiments and were intended to show the maximum fluorescence as the fluorophore and quencher should be locked at their maximum distance apart. We noticed however that as our triggers concentration increase the fluorescence reached a plateau at lower level than the NC for each switch. We hypothesised that as the NCs went right up to the fluorophore, compared to the triggers where there is a 6nt gap, the difference was due to the change in local chemical environment of the fluorophore rather than the distance from the quencher. We therefore designed 1*1* and 2*1* triggers fusions of two trigger complementary to the homo sym and hetero sym switches respectively. These should bind non-cooperatively but unlike the original NCs these do not approach any further towards the fluorophore than the normal 14nt triggers.
The sequence of all our ssDNA strands using in our in vitro testing can be found on our SI page.
Testing our switches in vivo posed a unique set of challenges relating to plasmid design. Our ideal construct would allow us to characterise the relationship between the concentration of short RNA triggers and the translation rate of an mRNA containing our riboswitch. We decided to measure the amount of trigger RNA using the RNA aptamer Broccoli and the translation rate using the far-red fluorescent protein mKate2. This particular RFP variant was chosen for its high brightness and because it is spectrally resolvable from Broccoli fluorescence. After codon optimisation using IDT software it was run with our switches in the Salis RBS calculator. On the basis of the Salis results we changed the base sequence slightly to remove an unintended RBS-like sequence that was causing initiation around mKate2 Met12.
Both mKate2 and Broccoli fluorescence measurements are qualitative in that they cannot be easily converted to an absolute number of fluorophores but we will relate both to the growth rate of the cells by dividing by the OD in a plate reader or by relating to cell volume in single cell measurements.
Broccoli was added to our Triggers as a 3’ fusion within the F30 RNA scaffold, which is based upon the highly stable viral ϕ29 three-way junction RNA motif, to reduce the chance of misfolding. The 2nt linker between this structure and the trigger we manually changed individually for each trigger by running multiple variations of the join sequence in Nupack and checking for correct folding. Broccoli within this scaffold is now referred to as F30(Bro).
After the F30(Bro) we included a longer AU rich linker then a small synthetic terminator. The linker was intentionally long and AU-rich because we wanted to ensure that if RNA degradation did occur we did not end up with free Broccoli and untagged trigger. This would separate the link between Broccoli fluorescence and trigger concentration. The terminator sequence ends the transcription of our trigger transcript so they are not excessively long and to prevent transcription read-through into other parts of our plasmid.
The main mRNA degradation pathway in E. coli (assumed to be the dominant pathway for our trigger degradation as it does not resemble either a tRNA or an rRNA) involves intramolecular cleavage by RNaseE, followed by 3’ exonuclease activity. Prior to this the mRNA is protected by the terminator hairpin and the lack of 5’ exonucleases. RNaseE cleavage 3’ to the F30(Bro) would lead to exonuclease digestion of both it and the Trigger whilst cleavage between the Broccoli and the trigger would lead to F30(Bro) without attached trigger.
We initially intended to use a version of Broccoli with four repeated Broccoli aptamer units, F30(2xdBro), that would be four times as bright as a single Broccoli unit. This was not used in the end as our designs were too repetitive for DNA synthesis.
1- Reporters
We put both our trigger-F30(Bro) transcript and switch-mKate2 transcript under inducible promoters rather than constitutive promoters for two main reasons:
-
The expression of artificial genes can negatively impact cell survival and growth rate. As we would like our cells to be happy as possible before testing, the genes not be transcribed until we induce expression at the start of an experiment.
-
With inducible expression we can alter the amount of trigger relatively to that of the switch and measure the result on mKate2 expression. The trigger/switch ratio can be used to measure of the cooperativity our system.
Our initial intention was to fix the induction of the switch-mKate2 during an experiment at an intermediate level whilst the expression of the triggers is varied by several orders of magnitude.
We used the pBAD arabinose-inducible promoter for the triggers, due to its high dynamic range and the pTrc promoter, a hybrid of the lac promoter and trp promoter, for the switch. The Trc promoter is regulated by lacI, like the lac promoter, but is less leaky. Without the repressor lacI, the pTrc promoter is constitutive, whilst the pBAD promoter requires its regulator araC for full activity.
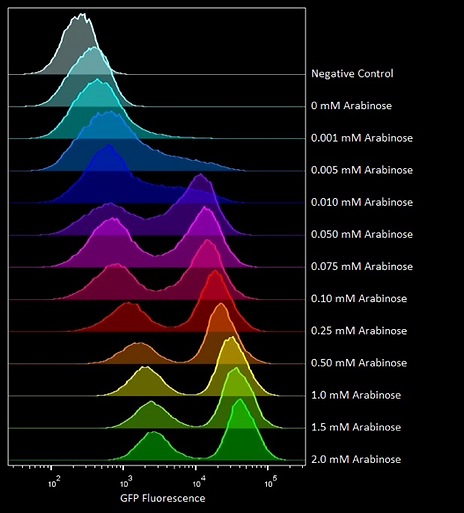
One major issue is that pBAD displays bimodal distribution with respect to arabinose concentration. This means that increasing arabinose concentration causes an increase in a population of fully induced cells relative to a population of mostly uninduced cells rather than a homogenous population of cells that all increase their expression. This means that when we measure the fluorescence of our system on a population basis, i.e. on a plate reader, this will actually represent an average measurement of two groups of cells with different trigger/switch ratios. Despite this the pBAD promoter was used because of its high expression ratio and we used microscopy/flow cytometry to measure trigger/switch ratio on a single cell basis.
An example of a bimodal distribution with pBAD can be seen in flow cytometry by Oxford iGEM in 2016:
2- Promoters
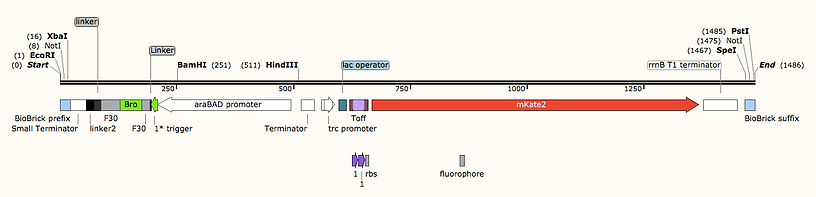
Parts were constructed with both the switch-mKate2 transcript, expressed from the pTrc promoter and Trigger-F30(Broccoli) transcript, divergently expressed from the pBAD promoter. Divergent expression should mean that any backwards activity of the promoter should result in a nonsense sequence but a terminator was also included between the two, in the majority of our constructs, to reduce the chance of readthrough.
An example test part (homo sym 8) ordered in a Twist Kanamycin-resistance plasmids:
The mKate2 gene was codon optimised for E. coli by IDT software then underwent a few adjustments by hand to improve DNA synthesis. Biobrick prefix and suffix segments were placed at the ends of our parts to aid transfer into other vectors in future applications. We also included a HindIII site between the pTrc-Switch-mKate and pBAD-Trigger-F30(Bro) should these want to be used separately.
Each test construct, with both switch-mKate2 transcript and Trigger-F30(Bro) transcript components, was synthesised by Twist bioscience and delivered in a plasmid with a ColE1 origin and kanamycin resistance gene. As both promoters require co-expression of their repressor proteins (lacI and araC) for regulated expression, the genomic copy being insufficient, we ordered these as separate genes under constitutive expression and planned to ligate them both expressed divergently into a plasmid with a compatible p15A origin with a different resistance gene. Due to time constraints, we did not manage to carry this out so we cotransformed with the commercial expression vector pBAD33 that expresses araC and contains a compatible p15A origin.
It is not standard practice to express the transcription factor proteins on a separate plasmid to the promoters they are controlling as there may be issues relating to the different copy numbers between the two plasmids. (500-700 per cell for the test constructs with the ColE1 origin but only around 15 per cell for the pBAD33 plasmid). However, we felt that putting the repressors on different plasmids was necessary as the short timescale of the project required us to eliminate cloning steps.
The use of pBAD33 only and not any plasmid containing lacI means that the pTrc promoter controlling the switch-mKate2 is essentially constitutive and not regulated by lactose or IPTG as we initially intended. This means that E. coli transformed with our repressive switch designs will have constitutive mKate2 expression in the absence of arabinose, however if our activatory designs operate correctly they will only express mKate2 if arabinose is added. We estimate that its unregulated level is appropriate for switching to have a significant impact whilst not excessively draining cell resources.
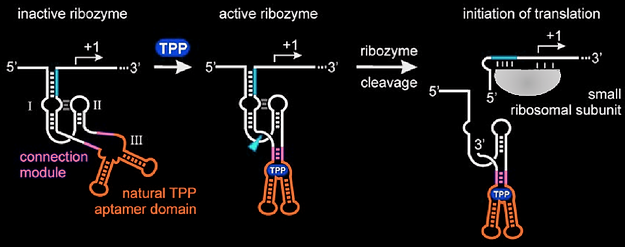
Designing plasmids for our heterotropic switches, which have two different triggers, in vivo, proved particularly challenging. If both triggers were expressed as one transcript, with F30(Bro) then they would not operate as separate molecules which is required for the design. However, if they were expressed from different promoters then it would be difficult to establish the relative concentration of each of them and use of a copy of the pBAD promoter for each Trigger exceeded our maximum part size. We settled upon a solution whereby the two triggers were expressed as a single transcript from the same promoter but separated by a TPP-inducible Hammerhead ribozyme. A hammerhead ribozyme an RNA enzyme able to catalyse its own self cleavage. This version is responsive to the intracellular concentration of thiamine pyrophosphate (TPP). Hartig et al found that in the intrinsic level of TPP biosynthesis by synthesis-proficient cells was insufficient for ribozyme stimulation. Stimulation could be induced by addition of thiamine to the external medium (that presumably is efficiently imported into the cells and converted to TPP by thiamine kinase and thiamine phosphate kinase). Our experimental plan was therefore to test the switch with by adding arabinose both with and without thiamine. Thiamine should convert the heterotropic switch from non-cooperative behaviour as both trigger sequences are part of the same molecule to cooperative behaviour as the transcript is cleaved between the two triggers. As both triggers are from the same transcript they can be expressed from one promoter, have one broccoli tag and should be equimolar. One issue with this design is that one trigger sequence ends up at the 5’ end of a piece of RNA whilst the other is in the middle of a piece of RNA proceeded by part of the cleaved ribozyme.
The sequences of all of our plasmid inserts can be found on our SI page.
3- Plasmids
References
Nupack:
-
J. N. Zadeh, C. D. Steenberg, J. S. Bois, B. R. Wolfe, M. B. Pierce, A. R. Khan, R. M. Dirks, N. A. Pierce. NUPACK: analysis and design of nucleic acid systems. J Comput Chem, 32:170–173, 2011.
-
R. M. Dirks, J. S. Bois, J. M. Schaeffer, E. Winfree, and N. A. Pierce. Thermodynamic analysis of interacting nucleic acid strands. SIAM Rev, 49:65-88, 2007.
-
R. M. Dirks and N. A. Pierce. An algorithm for computing nucleic acid base-pairing probabilities including pseudoknots. J Comput Chem, 25:1295-1304, 2004.
-
R. M. Dirks and N. A. Pierce. A partition function algorithm for nucleic acid secondary structure including pseudoknots. J Comput Chem, 24:1664-1677, 2003.
-
B. R. Wolfe, N. J. Porubsky, J. N. Zadeh, R. M. Dirks, and N. A. Pierce. Constrained multistate sequence design for nucleic acid reaction pathway engineering. J Am Chem Soc, 139:3134-3144, 2017.
-
B. R. Wolfe and N. A. Pierce. Sequence design for a test tube of interacting nucleic acid strands. ACS Synth Biol, 4:1086-1100, 2015.
-
J. N. Zadeh, B. R. Wolfe, and N. A. Pierce. Nucleic acid sequence design via efficient ensemble defect optimization. J Comput Chem, 32:439–452, 2011.
-
R. M. Dirks, M. Lin, E. Winfree, and N. A. Pierce. Paradigms for computational nucleic acid design. Nucl Acids Res, 32:1392-1403, 2004.
Salis:
-
Espah Borujeni, A., Channarasappa, A. and Salis, H. (2013). Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Research, 42(4), pp.2646-2659.
-
Salis, H., Mirsky, E. and Voigt, C. (2009). Automated design of synthetic ribosome binding sites to control protein expression. Nature Biotechnology, 27(10), pp.946-950.
RNaseE:
-
Mackie, G. (2012). RNase E: at the interface of bacterial RNA processing and decay. Nature Reviews Microbiology, 11(1), pp.45-57.
Promoters:
-
Balzer, S., Kucharova, V., Megerle, J., Lale, R., Brautaset, T. and Valla, S. (2013). A comparative analysis of the properties of regulated promoter systems commonly used for recombinant gene expression in Escherichia coli. Microbial Cell Factories, 12(1), p.26.
Brocolli:
-
Filonov, G. and Jaffrey, S. (2016). RNA Imaging with Dimeric Broccoli in Live Bacterial and Mammalian Cells. Current Protocols in Chemical Biology, pp.1-28.
Ribozymes:
-
Wieland, M., Benz, A., Klauser, B. and Hartig, J. (2009). Artificial Ribozyme Switches Containing Natural Riboswitch Aptamer Domains. Angewandte Chemie International Edition, 48(15), pp.2715-2718.
Our switches were tested in the model organism E. coli DH5α using fluorescence reporters. This common cloning strain was used rather than an Rnase-deficient mutant because we wanted to test whether our switches worked in an organism with normal RNA metabolism. If our switches operated within this context we would be more confident that they could be applied to other organisms that lack Rnase-free mutants.
Adapted from Hartif et al.

Oligo with 5' Fluorophore
Oligo with 3'
Quencher
Switch closed
∴Fl and Q close
∴Fluorescence is low

Switch opened
∴Fl and Q far apart
∴Fluorescence is high
Oligo with 5' Fluorophore
Switch closed
∴RBS inaccessible
∴Mimic cannot bind
∴Fl and Q far apart
∴Fluorescence is high
Switch opened
∴RBS accessible
∴Mimic can bind
∴Fl and Q close
∴Fluorescence is low
"Mimic" with 5' Fluorophore
Oligo with 3' Quencher
"Mimic" with 5' Fluorophore
Oligo with 3' Quencher
Oligo with 3' Quencher
Trigger x2
Trigger x2
Computational design
This page assumes a high level of biochemistry knowledge and is intended to help you understand the decisions we made in order to replicate and improve upon our designs. For a simplified description of our project please see our project page. For descriptions of all our designs see our activatory and repressive design pages.
Our riboswitch designs were made to regulate the expression of a downstream gene by using the RNA folding to control access to a ribosome binding site (RBS). Some were designed to open/close cooperatively in response to the addition of two or more copies of complementary trigger strands whilst others were intended to act as experimental controls or to help us understand various aspects of cooperativity.
For the purposes of this project, our final construct, that we will test in E. coli cells, will be a synthetic riboswitch controlling the fluorescent reporter gene mKate2 (a version of RFP) expressed from a version of the lac promoter. This reporter represents any gene that you want to be controlled by the switch. The trigger strand for this switch will be expressed from the same plasmid from the arabinose-dependent pBAD promoter. These switches will also be tested as molecular beacons in vitro.
All our riboswitch designs were made using the software Nupack. When given a desired structure and lengths of strands involved in forming that structure Nupack can generate a set of sequences that are able to fulfil the structure constraints. We imposed additional sequence constraints on these designs to dictate the location of the RBS.
For instance, to generate the sequences of our homotropic symmetrical activatory switch we used the code:
# parameters for NUPACK design
material = rna
temperature = 37
dangles = none
trials = 5
# target structure defined in dot-parens-plus notation
structure apt = ..............
structure Tact = .. ...... (((((((((((((( (((((((((((((( ...... ........ .......... .......... .......... +))))))))))))))+))))))))))))))
structure Toff = .. (((((( ((............ ............)) )))))) ........ .......... .......... ..........
# define sequence domains
domain end1 = N2
domain stem1 = N6
domain bs= N14
domain stem2= AGA GGA
domain end2= GA N6
domain gene= ATGGTGAGCGAGCTGATTAAGGAGAACATG
# thread domains onto strands
strand switch1 = end1 stem1 bs bs stem2 end2 gene
strand apt1 = bs*
# thread strands onto target structures
Tact.seq = switch1 apt1 apt1
Toff.seq = switch1
apt.seq = apt1
# prevent sequence patterns
prevent = AAAA, CCCC, GGGG, TTTT, CCGG, GGCC, CGCG, GCGC, KKKKKK, MMMMMM, RRRRRR, SSSSSS, WWWWWW, YYYYYY
Tact is the structure of our switch in the translationally active state, Toff is the structure in the translationally inactive state and apt is the structure of our trigger. Each dot represents an unpaired nucleotide, whilst brackets represent nucleotides in a base pair.
This code resulted in the sequences:
Switch: GGUCCUCUACAAGAAUAGGAGUACAAGAAUAGGAGUAGAGGAGAUACGAUAUGGUGAGCG
AGCUGAUUAAGGAGAACAUG
Trigger: ACUCCUAUUCUUGU
In the Toff state this structure has two nucleotides unpaired at the 5' end followed by a half of a stem of 8nt, a loop of 24nt and then other part of the hairpin. The RBS is in the second bit of the hairpin and is sticking out by two nucleotides. We used the RBS sequence used in the Toehold switches of Yin et al.
Our full Nupack code can be found on our SI page.
Designs were made in Nupack as RNA at 37°C as they are initially intended to be tested in E. coli which, as a commensal gut bacterium, grows best at this temperature as it is close to human body temperature. The first 10 codons of our fluorescent reporter gene mKate2 was included in the design in order to prevent any misfolding events.
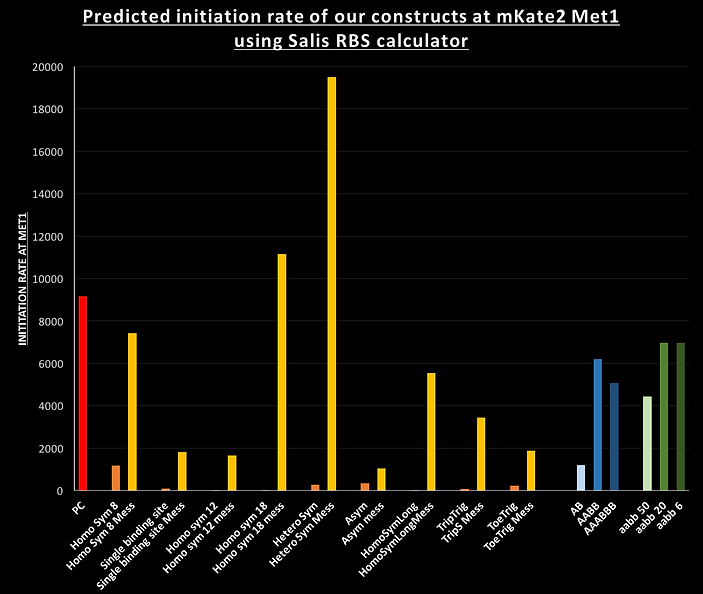
The designs were also optimised with the help of the Salis RBS calculator. When given a sequence of an mRNA this software can predict the translation initiation rate at every possible initiator codon based upon local mRNA structure. For our activatory designs we compared the initiation rate of our constructs in the Toff state, where the stem of the hairpin should hide the RBS, to a similar version (named "mess") where the sequence of first part of the hairpin was replaced with the second part i.e. preventing any hairpin formation whilst keeping the sequence as similar as possible. The Salis RBS calculator predicted the following initiation rates (in proportional units) for mKate2 at the methionine 1:
The repressive AB, AABB and AAABBB switches were particularly difficult as the translation from the RBS in the loop of the design leads to expression of a protein with an N-terminal peptide. We had to optimise the designs using Nupack and the Salis RBS calculator whilst keeping these peptides, on all three designs, as hydrophilic as possible. Hydrophobic peptides would have a tendency for the protein to misfold as it is being translated.

Trigger RNAs
mKate2 mRNA
Riboswitch
Triggers bind to riboswitch to affect translation
Translation
Transcription
Transcription
DNA
mKate2 gene
Trigger sequence
pBAD promoter
lac promoter
mKate2 protein
Riboswitch
RNA
Protein

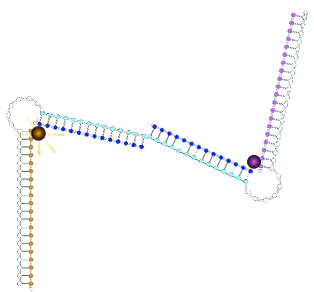
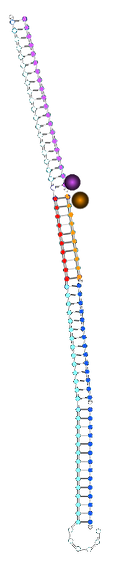
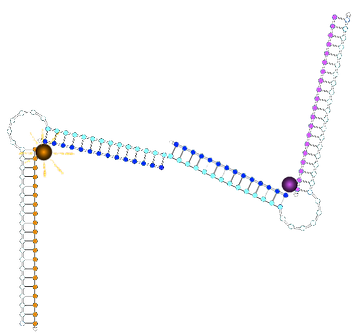
State with two 1* triggers bound:
State with original non-cooperative control strand:
State with new non-cooperative strand 1*1*:
ssDNA on the 3' side of fluorophore
dsDNA on the 3' side of fluorophore
∴environment different